
GtoPdb is requesting financial support from commercial users. Please see our sustainability page for more information.
Voltage-gated calcium channels (CaV): Introduction

INTRODUCTION
The family of voltage-gated calcium channels serve as the key transducers of cell surface membrane potential changes into local intracellular Ca2+ transients that initiate many different physiological events. There are ten members of the voltage-gated Ca2+ channel family that have been characterized in mammals, and they serve distinct roles in cellular signal transduction. This article introduces the molecular relationships and physiological functions of these Ca2+ channel proteins and provides background information on their molecular, genetic, physiological, and pharmacological properties.
Voltage-gated Ca2+ channels mediate Ca2+ influx in response to membrane depolarization and regulate intracellular processes such as contraction, secretion, neurotransmission, and gene expression in many different cell types [8,62]. Their activity is essential to couple electrical signals in the cell surface to physiological events in cells. They are members of a gene superfamily of transmembrane ion channel proteins that includes voltage-gated potassium and sodium channels [60]. This article presents an introduction to their biochemical, molecular, and genetic properties, their physiological roles, and their pharmacological significance, as an introduction to the comprehensive information on each member of the Ca2+ channel family in the Ion Channel Database.
Calcium Channel Subunit Architecture
The Ca2+ channels that have been well-characterized biochemically are complex proteins composed of four or five distinct subunits (Figure 1A; [8,49,51]). The α1 subunit of 190-250 kDα is the largest subunit and it incorporates the conduction pore, the voltage sensor and gating apparatus, and most of the known sites of channel regulation by second messengers, drugs, and toxins. An intracellular β subunit, a transmembrane, disulfide-linked α2δ subunit complex, and a transmembrane γ subunit are also components of CaV1.1 Ca2+ channels based on early biochemical studies [51].
The three-dimensional architecture of the skeletal muscle CaV1.1 (Figure 1B) [52,58-59,63] and CaV3.1 [64] channels has been elucidated at 3-4Å by cryo-electron microscopy. Remarkably, the overall organization of subunits within the complex closely resembles the model inferred from the original biochemical studies of the purified CaV1.1 channel complex [51]. The pore-forming α1 subunit is the central transmembrane component, with 24 transmembrane segments surrounding a central pore. The β subunit is located on the cytoplasmic side of the complex interacting primarily with the intracellular surfaces of domains I and II of the α1 subunit. The γ subunit is in a transmembrane position, with its four transmembrane segments interacting primarily with domain IV of the α1 subunit. The proteolytically processed, disulfide-linked α2δ subunit is in an extracellular position, interacting primarily with the extracellular surface of domains I-III of the α1 subunit. It is associated with the membrane through the δ subunit, but the transmembrane attachment is not shown clearly in the available cryo-EM structures [59]. Increasing evidence indicates that the α2δ subunit is further posttranslationally processed by proteolytic cleavage of its C-terminal transmembrane segment and attachment of a glycosylphosphatidylinositol anchor, which may be crucial for insertion into the plasma membrane at synaptic active zones and for recycling through endosomes for return to the plasma membrane [14].

Figure 1. Subunit architecture of calcium channels. Left. The subunit architecture of the skeletal muscle CaV1.1 channel is depicted as described in original biochemical studies [51]. P, protein phosphorylation. Curvy lines, glycosylation. Right. The subunit architecture of the skeletal muscle CaV1.1 channel is depicted as revealed in cryo-electron microscopy studies [59]. The different subdomains of the α2δ-1 subunit are shown in different colors: Von Willebrand Factor domain (green), Cache domain 1 (brown), and Cache domain 2 (purple). CTD, C-terminal domain.
Like the α subunits of sodium channels, the α1 subunit of voltage-gated Ca2+ channels is organised in four homologous domains (I--IV) with six transmembrane segments (S1-S6) in each (Figure 2A). The S4 segment contains the gating charges, which sense changes in the electric field and initiate conformational changes that open the pore. The pore loop between transmembrane segments S5 and S6 in each domain determines ion conductance and selectivity. Changes of only three amino acids in the pore loops in domains I, III, and IV will convert a sodium channel to Ca2+ selectivity [21]. Although auxiliary subunits modulate the properties of the channel complex, the pharmacological and electrophysiological diversity of Ca2+ channels arise primarily from the existence of multiple α1 subunits [24].
Calcium Channel Selectivity Filter at Atomic Resolution
Based on the amino acid sequences of P-loops of human Ca2+ channels [21], a Ca2+-selective ion selectivity filter was constructed in the bacterial CaV channel construct CaVAb by substitution of three negatively charged amino acid residues on the outer end of the selectivity filter [53]. This small molecular change does not change the conformation of the selectivity filter at all (Figure 2B, left), but it is sufficient to change the ion selectivity from strongly Na+-preferring (PCa/PNa ~ 0.03) to strongly Ca2+-preferring (PCa/PNa ~ 400) with an overall 12,000-fold difference in selectivity [53]. The additional negative charges create a series of Ca2+ binding sites across the membrane, as observed by x-ray crystallography [53]; Figure 2B, right). Sequential occupancy of these negatively charged coordination sites by Ca2+ yields rapid and selective Ca2+ conductance.
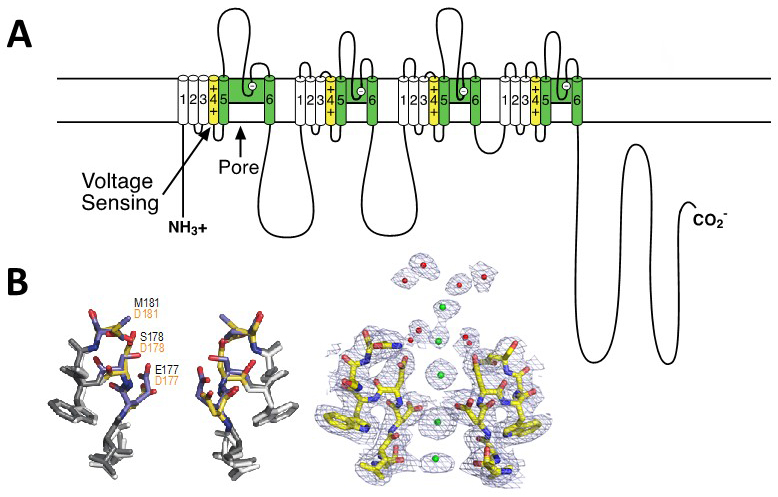
Figure 2. Structure of calcium channels. A. A transmembrane folding diagram of the voltage-gated calcium channel α11.1 subunit. Transmembrane segments S1-S4 form the voltage-sensing module. The S4 segments with their positive gating charges are highlighted in yellow. Transmembrane segments S5 and S6 and the P loop between them are highlighted in green. B. Structural basis for selective calcium conductance. Left. Superimposed high-resolution images of the ion selectivity filters of the bacterial sodium channel NaVAb and its Ca2+-selective derivative CaVAb. Native amino acid residues of NaVAb are in black; substituted amino acid residues in CaVAb are in red. Right. High-resolution structure of the calcium selectivity filter in CaVAb [53]. Amino acid residues of T1775 to D181 in stick representation. Green, calcium ions with electron density illustrated in mesh. Red, ordered water molecules.
Ca2+ Currents
Ca2+ currents recorded in different cell types have diverse physiological and pharmacological properties, and an alphabetical nomenclature evolved for the distinct classes of Ca2+ currents [46] [55]). L-type Ca2+ currents typically require a strong depolarisation for activation, are long-lasting, and are blocked by the organic L-type calcium antagonists, including dihydropyridines, phenylalkylamines, and benzothiazepines [32,43]. They are the main Ca2+ currents recorded in muscle and endocrine cells, where they initiate contraction and secretion. A subset of L-type Ca2+ currents activating at more negative voltages also are present in neurons and cardiac pacemaker cells. N-type, P/Q-type, and R-type Ca2+ currents also require strong depolarisation for activation [29,32,42]. They are relatively unaffected by L-type Ca2+ antagonist drugs but are blocked by specific polypeptide toxins from snail and spider venoms [33]. They are expressed primarily in neurons, where they initiate neurotransmission at most fast synapses and also mediate Ca2+ entry into cell bodies and dendrites. T-type Ca2+ currents are activated by small depolarisations from resting and are transient [6]
Ca2+ Channel Classification and Nomenclature
Mammalian α1 subunits are encoded by ten distinct genes. Historically, various names had been given to the corresponding gene products, giving rise to distinct and sometimes confusing nomenclatures. In 1994, a unified, but arbitrary nomenclature was adopted in which α1 subunits were referred to as α1S for the original skeletal muscle isoform and α1A through α1E for those discovered subsequently [3]. In 2000, a rational nomenclature was adopted [13] based on the well-defined potassium channel nomenclature [10]. Ca2+ channels were named using the chemical symbol of the principal permeating ion (Ca) with the principal physiological regulator (voltage) indicated as a subscript (CaV). The numerical identifier corresponds to the CaV channel α1 subunit gene subfamily (1 to 3) and the order of discovery of the α1 subunit within that subfamily (1 through n). According to this nomenclature, the CaV1 subfamily (CaV1.1 to CaV1.4) includes channels containing α1S, α1C, α1D, and α1F subunits, which mediate L-type Ca2+ currents (Table 1). The CaV2 subfamily (CaV2.1 to CaV2.3) includes channels containing α1A, α1B, and α1E, which mediate P/Q-type, N-type, and R-type Ca2+ currents, respectively (Table 1). The CaV3 subfamily (CaV3.1 to CaV3.3) includes channels containing α1G, α1H, and α1I, which mediate T-type Ca2+ currents (Table 1).
TABLE 1. Physiological function and pharmacology of calcium channels.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* Here we list only the primary localisations and the standard antagonists most widely used in research. Much more detail is given in the Ion Channel Database.
The complete amino acid sequences of these α1 subunits are more than 70% identical within a subfamily but less than 40% identical among the three subfamilies. These family relationships are illustrated for the conserved transmembrane and pore domains in Figure 3. Division of Ca2+ channels into these three families is phylogenetically ancient, as one representative of each is found in the C. elegans genome. Consequently, the genes for the different α1 subunits have become widely dispersed in the genome and even the most closely related members of the family are not clustered on single chromosomes in mammals.

Figure 3. Sequence similarity of voltage-gated calcium channel α1 subunits. Comparison of the amino acid sequence similarity of mammalian calcium channels. Only the membrane-spanning regions and pore loops are compared. All sequence pairs were aligned and compared, which led to the clear separation of the three subfamilies (CaV1, CaV2, CaV3) that have internal sequence identity of >80%. Consensus sequences were defined for all three families and compared to one another, yielding inter-family sequence identity of 52% (CaV1 vs. CaV2) and 28% (CaV3 vs. CaV1 or CaV2).
Ca2+ Channel Interacting Proteins
Ca2+ channels are regulated by transient interactions with G protein βγ subunits, SNARE proteins, calmodulin and related Ca2+ sensor proteins, and phosphorylation by several protein kinases [7,9,24,31] [44,61]. In addition to these transient protein-protein interactions, Ca2+ channel expression and localization are regulated by stable interactions with members of the RGK-family of Ras-like GTP-binding proteins, like Rad and Rem. They bind to the intracellular CaVβ subunit and reduce CaV channel expression [4]. Recent work on Rad indicates that its phosphorylation by the β-adrenergic/protein kinase A signaling pathway contributes importantly to up-regulation of the activity of Cav1.2 channels in the heart in response to stress and exercise [28]. RIM proteins bind to a PDX domain on the C-terminal of CaV2 channels and anchor them at presynaptic active zones [20,26]. They also modulate CaV1 and CaV2 channel activity through their binding to CaVβ-subunits [17,27,36]. The adaptor protein STAC3 binds to Cav1.1 and has been identified as essential component to link Cav1.1 and RyR1 for skeletal muscle EC coupling [16,25,41]. STAC proteins also regulate Ca2+/calmodulin-dependent feedback of L-type Ca2+ channels [5,40]. Binding of α-actinin to the C-terminus of Cav1.2 supports its surface localization and postsynaptic targeting in neurons [19]. It is likely that additional Ca2+ channel interacting proteins will be discovered that regulate channel expression, localization, and function [57].
Ca2+ Channel Molecular Pharmacology
The pharmacology of the three subfamilies of Ca2+ channels is quite distinct. The CaV1 channels are the molecular targets of the organic Ca2+ channel blockers used widely in the therapy of cardiovascular diseases. These drugs are thought to act at three separate, but allosterically coupled, receptor sites (Table 1; reviewed in Glossmann and Striessnig, 1990 [18]). A combination of photoaffinity labeling, site-directed mutagenesis, and molecular modeling provided an initial view of the location of the drug receptor sites for dihydropyridines, phenylalkylamines, and diltiazem on the CaV1.2 channel protein
Structural studies of the Cav1.1 channel from mammalian skeletal muscle have also revealed the binding sites for the three classes of Ca2+ antagonist drugs that act on Cav1 channels (Figure 4, right [63]). The receptor sites for verapamil and diltiazem closely resemble those found for CavAb. On the other hand, the receptor site for dihydropyridines is slightly, but significantly, different. Amlodipine bound to CavAb lies approximately two helical turns on the extracellular side of nifedipine bound to Cav1.1, at the outer edge of the dihydropyridine receptor site defined by mutagenesis of mammalian Cav1.2 channels [63]. This structural difference likely reflects differences in amino acid sequence in this region of the two Cav channels. Nevertheless, the mechanistic distinction between phenylalkylamines and benzothiazepines as direct pore blockers and dihydropyridines as indirect allosteric inhibitors is retained in these two distantly related Cav channel proteins.

Figure 4. Structure of the drug receptor sites in voltage-gated Ca2+ channels. Left: Top. Top view of the model Ca2+ channel CavAb. Voltage-sensing domains in blue; pore domain in gray. Ca2+ is bound in the central pore (red), and amlodipine is bound to its receptor site on the outer surface of the pore domain. Bottom. Side view. A cross section of CavAb with verapamil bound in the central Cavity in the pore, just below the ion selectivity filter. Right: The position of the pore domain-forming S5 and S6 helices within the rabbit Cav1.1 channel complex (rCav1.1) is shown on the left. The insets show the interaction sites for various Ca2+-channel blockers nifedipine, verapamil and diltiazem as well as the Ca2+-channel activator BAY K 8644 (from Wu et al., 2019, [63]).
The CaV2 subfamily of Ca2+ channels is relatively insensitive to dihydropyridine Ca2+ channel blockers; rather, these Ca2+ channels are specifically blocked with high affinity by peptide toxins from spiders and marine snails [33]. The CaV2.1 channels are blocked specifically by ω-agatoxin IVA from funnel web spider venom. The CaV2.2 channels are blocked specifically by ω-conotoxin GVIA and related cone snail toxins. Amongst the CaV family, CaV2.3 channels are blocked specifically by the synthetic peptide toxin SNX-482 derived from tarantula venom. These peptide toxins are potent blockers of synaptic transmission because of their specific effects on the CaV2 family of Ca2+ channels.
The CaV3 subfamily of Ca2+ channels is relatively insensitive to both the dihydropyridines that block CaV1 channels and the spider and cone snail toxins that block the CaV2 channels [37]. The organic Ca2+ channel blocker mibefradil is somewhat selective for T-type versus L-type Ca2+ currents (three- to ten-fold). The peptide kurtoxin inhibits the activation gating of CaV3.1 and CaV3.2 channels. Recently a number of pharmacological agents have been developed that selectively block T-type calcium currents, and these have been shown to be useful in models of neuropathic pain, epilepsy and psychiatric comorbidities (e.g., TTA-A2, TTA-P2, and Z944) [12,37]. Z944 has also proven efficacious in humans in an experimental model of neuropathic pain and central sensitization. The cryo-EM structures of human Ca v3.1 alone and in complex with Z944 have been reported recently [64]. For a complete listing of CaV3 inhibitors, please see the individual entries in the Ion Channel Database.
Ca2+ Channelopathies
Mutations in voltage-gated Ca2+ channels cause several forms of inherited disease [31]. Mutations in skeletal muscle CaV1.1 channels cause hypokalemic periodic paralysis [15,31,56]. Mutations that cause loss of voltage-dependent inactivation in cardiac and neuronal CaV1.2 channels cause Timothy Syndrome, a multi-faceted disease that includes cardiac arrhythmia, autism, and developmental abnormalities [30,47-48]. Mutations that cause gain of function in CaV2.1 channels cause migraine headache [39], and mutations that lead to missense substitutions and truncations as well as polyglutamine expansions in the large C-terminal domain of CaV2.1 cause spinocerebellar ataxia type 6 [34,65]. Loss of CaV1.3 function results in sinoatrial node dysfunction and deafness [1], whereas gain of function causes severe neurodevelopmental disorders (including autism) and primary aldosteronism [35]. Loss of function mutations of CaV1.4 channels prevent normal formation of synapses in photoreceptors and cause congenital stationary night blindness type 2 [2,50]. Gain of function mutations in other Cavs also cause disease, including epileptic encephalopathies (Cav2.3) [22], childhood-onset cerebellar ataxia (Cav3.1) [11] and primary aldosteronism (Cav3.2) [45]. More details on these channelopathies are given in the Ion Channel Database entries for each channel type.
References
1. Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nürnberg G, Ali A, Ahmad I, Sinnegger-Brauns MJ, Brandt N et al.. (2011) Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci, 14 (1): 77-84. [PMID:21131953]
2. Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. (1998) Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet, 19 (3): 264-7. [PMID:9662400]
3. Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Horne WA, Mori Y, Schwartz A, Snutch TP, Tanabe T et al.. (1994) The naming of voltage-gated calcium channels. Neuron, 13 (3): 505-6. [PMID:7917287]
4. Buraei Z, Yang J. (2015) Inhibition of Voltage-Gated Calcium Channels by RGK Proteins. Curr Mol Pharmacol, 8 (2): 180-7. [PMID:25966691]
5. Campiglio M, Costé de Bagneaux P, Ortner NJ, Tuluc P, Van Petegem F, Flucher BE. (2018) STAC proteins associate to the IQ domain of CaV1.2 and inhibit calcium-dependent inactivation. Proc Natl Acad Sci U S A, 115 (6): 1376-1381. [PMID:29363593]
6. Carbone E, Lux HD. (1984) A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature, 310 (5977): 501-2. [PMID:6087159]
7. Catterall WA. (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol, 16: 521-55. [PMID:11031246]
8. Catterall WA. (2011) Voltage-gated calcium channels. Cold Spring Harb Perspect Biol, 3 (8): a003947. [PMID:21746798]
9. Catterall WA. (2015) Regulation of Cardiac Calcium Channels in the Fight-or-Flight Response. Curr Mol Pharmacol, 8 (1): 12-21. [PMID:25966697]
10. Chandy KG, Gutman GA. (1993) Nomenclature for mammalian potassium channel genes. Trends Pharmacol Sci, 14 (12): 434. [PMID:8122319]
11. Chemin J, Siquier-Pernet K, Nicouleau M, Barcia G, Ahmad A, Medina-Cano D, Hanein S, Altin N, Hubert L, Bole-Feysot C et al.. (2018) De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain, 141 (7): 1998-2013. [PMID:29878067]
12. Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic-Todorovic V, Todorovic SM. (2011) TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol, 80 (5): 900-10. [PMID:21821734]
13. Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. (2000) Nomenclature of voltage-gated calcium channels. Neuron, 25 (3): 533-5. [PMID:10774722]
14. Ferron L, Kadurin I, Dolphin AC. (2018) Proteolytic maturation of α2δ controls the probability of synaptic vesicular release. Elife, 7. DOI: 10.7554/eLife.37507 [PMID:29916807]
15. Flucher BE. (2020) Skeletal muscle CaV1.1 channelopathies. Pflugers Arch, 472 (7): 739-754. [PMID:32222817]
16. Flucher BE, Campiglio M. (2019) STAC proteins: The missing link in skeletal muscle EC coupling and new regulators of calcium channel function. Biochim Biophys Acta Mol Cell Res, 1866 (7): 1101-1110. [PMID:30543836]
17. Gebhart M, Juhasz-Vedres G, Zuccotti A, Brandt N, Engel J, Trockenbacher A, Kaur G, Obermair GJ, Knipper M, Koschak A et al.. (2010) Modulation of Cav1.3 Ca2+ channel gating by Rab3 interacting molecule. Mol Cell Neurosci, 44 (3): 246-59. [PMID:20363327]
18. Glossmann H, Striessnig J. (1990) Molecular properties of calcium channels. Rev Physiol Biochem Pharmacol, 114: 1-105. [PMID:2155469]
19. Hall DD, Dai S, Tseng PY, Malik Z, Nguyen M, Matt L, Schnizler K, Shephard A, Mohapatra DP, Tsuruta F et al.. (2013) Competition between α-actinin and Ca²⁺-calmodulin controls surface retention of the L-type Ca²⁺ channel Ca(V)1.2. Neuron, 78 (3): 483-97. [PMID:23664615]
20. Han Y, Kaeser PS, Südhof TC, Schneggenburger R. (2011) RIM determines Ca²+ channel density and vesicle docking at the presynaptic active zone. Neuron, 69 (2): 304-16. [PMID:21262468]
21. Heinemann SH, Terlau H, Stühmer W, Imoto K, Numa S. (1992) Calcium channel characteristics conferred on the sodium channel by single mutations. Nature, 356 (6368): 441-3. [PMID:1313551]
22. Helbig KL, Lauerer RJ, Bahr JC, Souza IA, Myers CT, Uysal B, Schwarz N, Gandini MA, Huang S, Keren B et al.. (2018) De Novo Pathogenic Variants in CACNA1E Cause Developmental and Epileptic Encephalopathy with Contractures, Macrocephaly, and Dyskinesias. Am J Hum Genet, 103 (5): 666-678. [PMID:30343943]
23. Hockerman GH, Peterson BZ, Johnson BD, Catterall WA. (1997) Molecular determinants of drug binding and action on L-type calcium channels. Annu Rev Pharmacol Toxicol, 37: 361-96. [PMID:9131258]
24. Hofmann F, Lacinová L, Klugbauer N. (1999) Voltage-dependent calcium channels: from structure to function. Rev Physiol Biochem Pharmacol, 139: 33-87. [PMID:10453692]
25. Horstick EJ, Linsley JW, Dowling JJ, Hauser MA, McDonald KK, Ashley-Koch A, Saint-Amant L, Satish A, Cui WW, Zhou W et al.. (2013) Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat Commun, 4: 1952. [PMID:23736855]
26. Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC. (2011) RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell, 144 (2): 282-95. [PMID:21241895]
27. Kiyonaka S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, Beedle AM, Mori E, Hara Y, De Waard M et al.. (2007) RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci, 10 (6): 691-701. [PMID:17496890]
28. Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A et al.. (2020) Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature, 577 (7792): 695-700. [PMID:31969708]
29. Llinás R, Sugimori M, Hillman DE, Cherksey B. (1992) Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous system. Trends Neurosci, 15 (9): 351-5. [PMID:1382335]
30. Marcantoni A, Calorio C, Hidisoglu E, Chiantia G, Carbone E. (2020) Cav1.2 channelopathies causing autism: new hallmarks on Timothy syndrome. Pflugers Arch, 472 (7): 775-789. [PMID:32621084]
31. Nanou E, Catterall WA. (2018) Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron, 98 (3): 466-481. [PMID:29723500]
32. Nowycky MC, Fox AP, Tsien RW. (1985) Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature, 316 (6027): 440-3. [PMID:2410796]
33. Olivera BM, Miljanich GP, Ramachandran J, Adams ME. (1994) Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins. Annu Rev Biochem, 63: 823-67. [PMID:7979255]
34. Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M et al.. (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell, 87 (3): 543-52. [PMID:8898206]
35. Ortner NJ, Kaserer T, Copeland JN, Striessnig J. (2020) De novo CACNA1D Ca2+ channelopathies: clinical phenotypes and molecular mechanism. Pflugers Arch, 472 (7): 755-773. [PMID:32583268]
36. Ortner NJ, Pinggera A, Hofer NT, Siller A, Brandt N, Raffeiner A, Vilusic K, Lang I, Blum K, Obermair GJ et al.. (2020) RBP2 stabilizes slow Cav1.3 Ca2+ channel inactivation properties of cochlear inner hair cells. Pflugers Arch, 472 (1): 3-25. [PMID:31848688]
37. Perez-Reyes E. (2003) Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev, 83 (1): 117-61. [PMID:12506128]
38. Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. (1998) Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature, 391 (6670): 896-900. [PMID:9495342]
39. Pietrobon D, Striessnig J. (2003) Neurobiology of migraine. Nat Rev Neurosci, 4 (5): 386-98. [PMID:12728266]
40. Polster A, Dittmer PJ, Perni S, Bichraoui H, Sather WA, Beam KG. (2018) Stac Proteins Suppress Ca2+-Dependent Inactivation of Neuronal l-type Ca2+ Channels. J Neurosci, 38 (43): 9215-9227. [PMID:30201773]
41. Polster A, Perni S, Bichraoui H, Beam KG. (2015) Stac adaptor proteins regulate trafficking and function of muscle and neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A, 112 (2): 602-6. [PMID:25548159]
42. Randall A, Tsien RW. (1995) Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci, 15 (4): 2995-3012. [PMID:7722641]
43. Reuter H. (1979) Properties of two inward membrane currents in the heart. Annu Rev Physiol, 41: 413-24. [PMID:373598]
44. Reuter H. (1983) Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature, 301 (5901): 569-74. [PMID:6131381]
45. Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, Prasad ML, Goh G, Carling T, Juhlin CC et al.. (2015) Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife, 4: e06315. [PMID:25907736]
46. Snutch TP, Leonard JP, Gilbert MM, Lester HA, Davidson N. (1990) Rat brain expresses a heterogeneous family of calcium channels. Proc Natl Acad Sci USA, 87 (9): 3391-3395. [PMID:1692134]
47. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. (2005) Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci USA, 102 (23): 8089-8096; discussion 8086-8088. [PMID:15863612]
48. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K et al.. (2004) Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell, 119 (1): 19-31. [PMID:15454078]
49. Striessnig J. (1999) Pharmacology, structure and function of cardiac L-type Ca(2+) channels. Cell Physiol Biochem, 9 (4-5): 242-69. [PMID:10575201]
50. Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BH, Wutz K, Gutwillinger N, Rüther K, Drescher B et al.. (1998) An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet, 19 (3): 260-3. [PMID:9662399]
51. Takahashi M, Seagar MJ, Jones JF, Reber BF, Catterall WA. (1987) Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc Natl Acad Sci U S A, 84 (15): 5478-5482. [PMID:2440051]
52. Tang L, Gamal El-Din TM, Lenaeus MJ, Zheng N, Catterall WA. (2019) Structural Basis for Diltiazem Block of a Voltage-Gated Ca2+ Channel. Mol Pharmacol, 96 (4): 485-492. [PMID:31391290]
53. Tang L, Gamal El-Din TM, Payandeh J, Martinez GQ, Heard TM, Scheuer T, Zheng N, Catterall WA. (2014) Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature, 505 (7481): 56-61. [PMID:24270805]
54. Tang L, Gamal El-Din TM, Swanson TM, Pryde DC, Scheuer T, Zheng N, Catterall WA. (2016) Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nature, 537 (7618): 117-121. [PMID:27556947]
55. Tsien RW, Lipscombe D, Madison D, Bley K, Fox A. (1995) Reflections on Ca(2+)-channel diversity, 1988-1994. Trends Neurosci, 18 (2): 52-4. [PMID:7537405]
56. Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, Tristani-Firouzi M, Tawil R, Griggs RC, CINCH investigators. (2006) The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain, 129 (Pt 1): 8-17. [PMID:16195244]
57. Wang S, Cortes CJ. (2021) Interactions with PDZ proteins diversify voltage-gated calcium channel signaling. J Neurosci Res, 99 (1): 332-348. [PMID:32476168]
58. Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, Yan N. (2016) Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 Å resolution. Nature, 537 (7619): 191-196. [PMID:27580036]
59. Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M, Yan N. (2015) Structure of the voltage-gated calcium channel Cav1.1 complex. Science, 350 (6267): aad2395. [PMID:26680202]
60. Yu FH, Catterall WA. (2004) The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE, 2004 (253): re15. [PMID:15467096]
61. Zamponi GW. (2016) Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov, 15 (1): 19-34. [PMID:26542451]
62. Zamponi GW, Striessnig J, Koschak A, Dolphin AC. (2015) The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol Rev, 67 (4): 821-70. [PMID:26362469]
63. Zhao Y, Huang G, Wu J, Wu Q, Gao S, Yan Z, Lei J, Yan N. (2019) Molecular Basis for Ligand Modulation of a Mammalian Voltage-Gated Ca2+ Channel. Cell, 177 (6): 1495-1506.e12. [PMID:31150622]
64. Zhao Y, Huang G, Wu Q, Wu K, Li R, Lei J, Pan X, Yan N. (2019) Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature, 576 (7787): 492-497. [PMID:31766050]
65. Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet, 15 (1): 62-9. [PMID:8988170]




